Some search problems are MASSIVE! Could a triple store be used to make them easier? (Spoiler Alert: Yes!)…
I’ve been thinking about putting DNA nucleotide sequences in to MarkLogic server for comparison. By modelling a DNA sequence as a set of XML elements rather than one long string, MarkLogic’s universal index can be used to rapidly perform an exact match or contains search. This executes in sub second response time on a laptop.
The main problems with DNA analysis though are:-
- Sequences may not ‘line up’ – That is to say that is the first 8 characters of one sequence are AACTGCAG and the second sequence starts AAACTGCAG, we know as humans that sequence 1 is contained within sequence 2, with an offset of one character. Teaching a computer to spot this quickly is very difficult.
- Matching segments may have differing gaps – Sequence 1 may be AACT-GC-ACTG whereas sequence 2 may be AACT-AAGC-ACTG. Thus there’s an extra two basepairs in the ‘gap’ within a sequence
- Sequences may be reversed – AACTGCACTG may be reversed as GTCACGTCAA. If spliced ready for protein creation, this is still the same segment with the same effect, but it’s stored backwards in DNA
- Sequences may contain wildcards – Sequencing machines don’t always determine a base pair. Thus AACTG?ACTG – where the C isn’t determined – is not unusual.
- Performing comparisons – The computational complexity of performing a comparison is non trivial. For a simple percentage match against a database of millions of sequences you can be waiting a long time – and that will probably not be the most accurate comparison – the aim is to find a near match and compare the few candidates later
Reducing the search space
The problem is that a computer has no ‘prior knowledge’ of how a sequence may match others. Thus the search space is the entire database, and the compare space is the length of every single sequence, including the candidate one.
This is a very expensive algorithm in terms of complexity. If we can reduce the search space, we can reduce the complexity, making the algorithm faster.
The reason search engines are useful on the web is that I don’t have to go and check every page when I want to execute a query – I instead check an index.
Usually this index lists a set of terms, and the pages those terms appear in. Thus inverting the problem – hence the name reverse indexes.
I wondered if this pattern can be applied to DNA? Can we index DNA as a set of terms rather than individual letters? Perhaps this could help us answer many questions, and do so faster than traditional methods.
I can to the conclusion that candidate segments could be stored as a single entity rather than many letters. So instead of having to do 8 comparisons against AACTGCAC I instead, perhaps, compare against two entities ‘AACT’ and ‘GCAC’.
Perhaps these two segments exist in other sequences. Consider the following 5 sequences:-
1 AACTGCATTGCACCGA 2 AACTGCCGTGCACCGA 3 CAAACTGCCGTGCACCGA 4 AACTCGCGCGCCTCGA 5 AACTGGGGCGCCTCGA
The changes between sequences are shown in bold.
An indexing algorithm can be designed such that:-
- The new sequence is checked to see if it contains the largest segment currently in the database
- If it does not contain it, try the next longest sequence
- If it does contain it, process down and up the sequence to find other segment matches
- Create a Subject in the triple store for each sequence step, linking to the existing Subject for that particular segment match
- Whilst indexing, compute the similarity overall with other sequences that link to the same segments
This would in effect give you the below graph for all five sequences:-
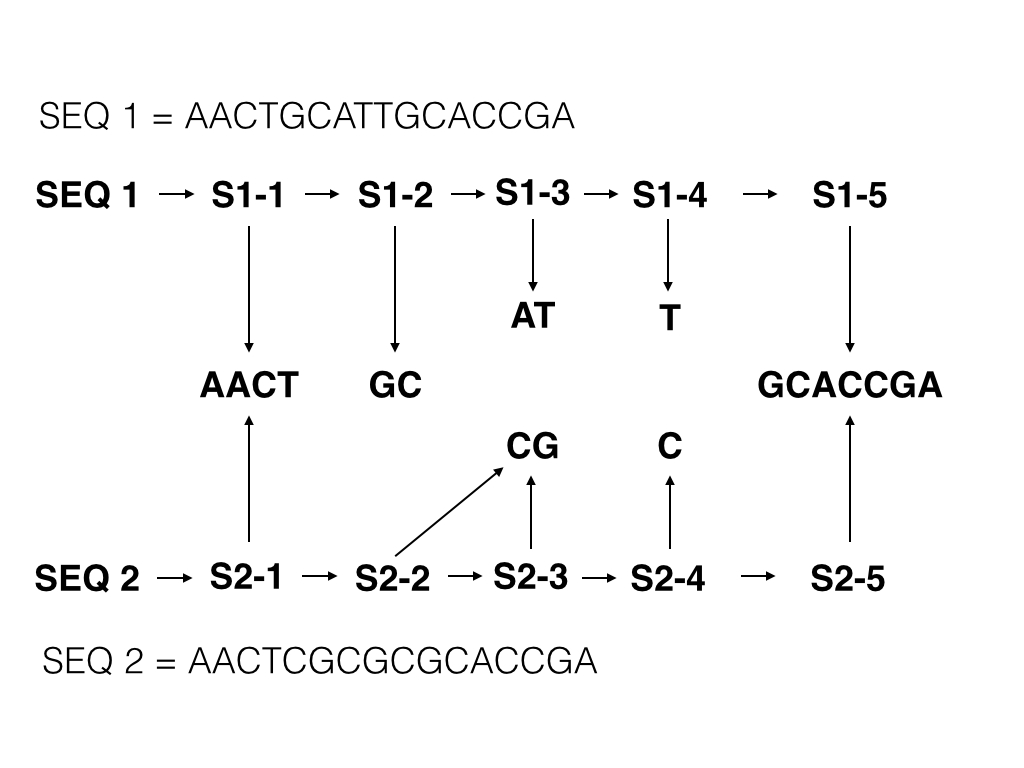
This solves the search space problem by breaking down a sequence of 16 distinct characters in to just 5 distinct Subject (each a segment of the original).
Other benefits of a graph model
Having a graph model produces a number of other benefits:-
- Non-aligned sequence analysis is easier, because we care about shared nodes no matter their position rather than trying to determine computationally a DNA alignment
- The ‘most similar’ sequences can be easily determined during indexing, because you can find other sequences with the same segments, and sum up the number of matches during indexing
- Search relevance (comparison score) is not calculated on the fly – each sequence would have a list of its most similar sequences. This is effectively a pre-filled ‘cache’ pattern. This makes comparison as easy as a lookup rather than a heavy duty computation job
- Other higher level biological entities, such as genes, can also be modelled as a sequence of segments, making their lookup easier after indexing (you could even pre-build a list of genes found in each sequence)
- The opposite half of a sequence (i.e. in the above first row, TTGACGTAACGTGGCT) can be modelled as a ‘sameAs’ relationship, making comparisons of those very quick indeed
- Other assertions can be modelled and tested. E.g. ‘Marker sequence X MARKS gene sequence Y’ or ‘Gene (Sequence) Q PRODUCES Protein (Sequence) R’
- The same method works for any linear construct that can be decomposed in to segments. In biology this includes protein amino acid chains, but could also be applied to project plans with interdependent tasks, or a list of multiple people’s activities or travels. (E.g. to find people who are often travelling to the same place, if not at the same time).
- Inferencing can be used to shortcut some of the relationships. E.g. skipping the ‘sequence -> segments <- gene’ relationships to generate ‘sequence CONTAINS gene’ triples.
Why has no one else thought of this?
They kind of have. There are a few people who have looked at graphs, but really to model facts about sequences (E.g. mathematical ‘distance’) rather than the sequences themselves. See Appendix A, [1] below for an example.
Next steps
The next steps are to try and mathematically compare the types of function a biologist may want to do, across two methods – the traditional brute force approach, and the above semantic sequence model approach.
This will give a good indication as to complexity of computing a comparison, and searching (computing relevance / or ‘well fit’) those comparisons and sequences.
A good semantic model can then be constructed and tested. I’m going to use MarkLogic Server for this as a) I know it well, and b) I can scale it out if needs be.
I’ve previously modelled DNA as element sequences in MarkLogic, so modelling them as triples in the same tool should provide a good comparison.
Appendix A: Previous research
[1] A Novel Model for DNA Sequence Similarity Analysis Based on Graph Theory
Xingqin Qi, Qin Wu, Yusen Zhang, Eddie Fuller, and Cun-Quan Zhang
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3204935/
This used a directed graph to hold weighted distances, not sequence information
